Инверсия пола 46 XY
OMIM 400044
Наша команда профессионалов ответит на ваши вопросы
Инверсия пола, 46,XY
Наличие женского фенотипа при нормальном мужском кариотипе характеризует XY-инверсию пола. Наиболее частой причиной данного нарушения формирования пола является синдром Свайера – это полная или «чистая» дисгенезия гонад при кариотипе 46,XY. Частота XY-дисгенезии гонад составляет 1 на 30000 человек. Больные имеют женский фенотип без признаков двойственности полового развития: феминное телосложение, развитые по женскому типу наружные половые органы, нормально развитую или гипоплазированную матку и маточные (фаллопиевы) трубы. Однако у пациентов с синдромом Свайера практически отсутствуют женские половые железы, которые в данном случае представлены дисгенетичными гонадами, представляющими собой соединительнотканные тяжи (стреки) с небольшими включениями железистой ткани, овариально-подобной стромы без фолликулов. Как правило, диагностирование синдрома Свайера происходит у девочек в пубертатный период, когда у них не происходит нормального полового развития. Причиной обращения к врачу при этом является задержка полового развития и отсутствие начала менструаций, реже наличие злокачественных новообразований, происходящих из дисгенетичных гонад. Так как дисгенетичные гонады подвержены озлокачествлению, показано их удаление в детстве или на момент постановки диагноза XY-дисгенезии гонад. После оперативного лечения пациенткам, как правило, еще в подростковом возрасте назначается заместительная гормональная терапия, чтобы достичь нормального развития вторичных половых признаков и предотвратить развитие остеопороза. У женщин с XY-дисгенезией гонад нет собственных яйцеклеток, однако в некоторых случаях она в состоянии выносить плод, полученный в программе ЭКО при оплодотворении донорской яйцеклетки сперматозоидами супруга.
Инверсия пола, 46,XY тип 1 (OMIM 400044)
Наиболее частой из известных причин «чистой» формы дисгенезии гонад 46,XY являются микроструктурные перестройки Y-хромосомы c утратой гена SRY (Sex-determining region Y), а также точковые мутации данного гена. У 10-15% больных с синдромом Свайера обнаруживают отсутствие локуса SRY. В большинстве случаев это обусловлено утратой фрагмента дистальной части короткого плеча Y-хромосомы (Yp11.3), вследствие X-Y транслокации. Еще у 10-15% пациентов с данным синдромом выявляют мутации гена SRY.
Ген SRY локализован на коротком плече Y хромосомы и кодирует транскрипционный фактор – белок, связывающийся с генами, определяющими развитие пола плода по мужскому типу. Мутации в гене SRY приводят к синтезу функционально неполноценного белка и к нарушению дифференцировки клеток Сертоли и формирования семенных канальцев в развивающихся бипотенциальных гонадах плода, что вызывает дисгенезию гонад и развитие остальных органов половой системы по женскому типу, несмотря на наличие Y-хромосомы в кариотипе.
Инверсия пола, 46,XYтип 2 (OMIM 300018)
Данный тип XY-инверсии пола обусловлен дупликаций гена NR0B1 (DAX-1). Ген NR0B1локализован на коротком плече Х хромосомы (локус Хp21.3). Кодируемый этим геном белок DAX-1 играет важную роль в развитии и функции некоторых органов эндокринной системы, в том числе и половых желез. Еще внутриутробно он контролирует активность генов, участвующих в формировании этих тканей, а в постнатальном периоде DAX-1 регулирует выработку в них гормонов. Белок DAX-1 оказывает дозо-зависимый эффект на органы эндокринной системы. Дупликация гена NR0B1, а также делеция располагающегося рядом с геном NR0B1 локуса, негативно-регулирующего его транскрипцию приводит к XY-инверсии пола, обусловленной XY-дисгенезией гонад часто сочетающейся с нарушением функции надпочечников. Точковые мутации этого гена у пациентов с кариотипом 46,XY вызывают нарушение развития тестикулярной ткани, приводят к дефициту маскулинизации. Мутации в этом гене также вызывают Х-сцепленную гипоплазию надпочечников, как у пациентов с кариотипом 46,ХХ так и 46,XY.
Инверсия пола, 46,XY тип 3 (OMIM 612965)
Инверсия пола, 46,XY тип 4 (OMIM 154230)
Эта форма XY-инверсии пола обусловлена делецией локуса 9p24.3. У пациенток отмечают нормально развитые по женскому типу наружные половые органы, нормально развитую или гипоплазированную матку, при гистологическом исследовании гонад обнаруживают наличие незрелой тестикулярной ткани, содержащей клетки Сертолли, и отсутствие зрелых половых клеток. Инверсия пола у данных пациентов, вероятно, обусловлена потерей одной из копий дозо-чувствительного гена, локализованного в данном локусе. Генами-кандидатами являются DMRT1 и DMRT2.
Инверсия пола, 46,XY тип 5 (OMIM 613080)
Данная аутосомно-рецессивная форма инверсии 46,XY обусловлена наличием мутаций в гене CBX2, расположенного на хромосоме 17 (локус 17q25). В 2009 году Байсон-Лаубер описал случай новорожденной девочки с кариотипом 46,XY, у которой в результате секвенированияв гене CBX2 были обнаружены две мутации (P98L и R443P). В результате исследований у девочки были обнаружены нормально развитые яичники, с наличием овариальной ткани и первичных фолликулов, а также влагалище и матка. Однако возраст еще был слишком мал, чтобы оценить ее фертильность и дальнейшее половое развитие.
Инверсия пола, 46,XY тип 6 (OMIM 613762)
XY-инверсия пола связана с наличием мутации в гетерозиготном состоянии в гене MAP3K1, расположенном в локусе 5q11.2. Пациентки с данной формой дисгенезии гонад имеют высокий рост, который, вероятно, обусловлен избыточной продукцией андрогенов, тяжевидные яичники, гипоплазированную матку, иногда наблюдается клиторомегалия.
Инверсия пола, 46,XY тип 7 (OMIM 233420)
Инверсия пола обусловлена наличием у пациенток мутаций в гомозиготном или компаунд-гетерозиготном состоянии в гене DHH, расположенного в локусе 12q13.12. У нескольких пациенток было описано наличие недоразвитой матки, также присутствовали фаллопиевы трубы и наблюдали полную форму ХY-дисгенезии гонад (тяжевидные гонады, которые часто озлокачествлялись).
Инверсия пола, 46,XYтип 8 (OMIM 614279)
Данный тип XY-инверсии пола обусловлен мутациями гена AKR1C2, лежащего в локусе 10p15, отвечающего за альтернативный путь синтеза дигидротестостерона. Мутации сцепленного с ним гена AKR1C4, который сегрегирует вместе с геном AKR1C2, могут влиять на выраженность фенотипических проявлений.
В Центре Молекулярной Генетики проводится молекулярный анализ ключевых генов, контролирующих дифференцировку пола, в частности выполняется секвенирование генов SRY и NR5A1 (SF1), а также с помощью количественного метода MLPA проводится поиск делеций и дупликаций генов SRY, NR5A1 (SF1), NR0B1 (DAX-1).
Инверсия пола 46 XX
OMIM 400045
Наша команда профессионалов ответит на ваши вопросы
46,ХХ инверсия пола характеризуется наличием мужского фенотипа (с полной или неполной маскулинизацией), наличием тестикулярной ткани при отсутствии в кариотипе Y-хромосомы. При этом данное нарушение формирования пола может быть обусловлено либо наличием синдрома «46,ХХ-мужчина» либо овотестикулярной формой нарушения формирования пола (истинный гермафродитизм). 46,ХХ инверсия пола может быть связана с наличием фрагмента Y-хромосомы и/или скрытого мозаицизма по Y-хромосоме (Y-позитивная форма) либо с аутосомными или Х-сцепленными мутациями (Y-негативная форма).
46,ХХ инверсия пола тип 1 (OMIM 400045)
В большинстве случаев ХХ-инверсия пола является результатом транслокации небольшого фрагмента короткого плеча Y-хромосомы, несущего ген SRY (OMIM 480000; Yp11.3), на Х-хромосому или аутосому. Нарушение формирования пола является врожденным состоянием, при котором наблюдается полное или частичное аномальное развитие и строение половых желез, внешних половых признаков, обусловленное аномалиями строения половых хромосом. У пациентов с истинным гермафродитизмом гистологически могут быть обнаружены как зрелые ткани яичников с фолликулами, так и яичек с семенными канальцами.
Ключевую роль в детерминации мужского пола и в дифференцировке яичек связана с геном SRY (Sex-determining region Y chromosome). Делеции или точковые мутации этого гена приводят к развитию «чистой» формы дисгенезии гонад при кариотипе 46,XY (синдром Свайера), тогда как его присутствие в геноме больных с 46,ХХ инверсией пола (синдром де ля Шапеля или синдром «46,ХХ-мужчина») обуславливает развитие по мужскому типу, а у больных с мозаицизмом по хромосоме Y, в том числе при синдроме Шерешевского-Тернера – с наличием и выраженностью маскулинизации и/или двойственного полового развития.
Ген SRY расположен на коротком плече Y-хромосомы в непосредственной близости к PAR1 региону, области гомологичной конъюгации хромосом Х и Y, происходящей в сперматогенезе. В процессе мейотического обмена между Х- и Y-хромосомами участок Y-хромосомы, содержащий ген SRY, может быть транслоцирован на Х-хромосому, что может привести к образованию сперматозоидов с перестроенными (дериватными) хромосомами: с Y-хромосомой, утратившей ген SRY, и Х-хромосомой, несущей этот ген. При оплодотворении такими гаметами яйцеклетки, соответственно, возможно рождение мужчин с кариотипом 46,ХХ с транслокацией гена SRY и женщин с кариотипом 46,XY, но с делецией гена SRY. Так, 85-90% больных с ХХ инверсией пола имеют в геноме небольшую часть короткого плеча Y-хромосомы, невидимую при стандартном цитогенетическом исследовании. Последовательности Y-хромосомы у таких больных транслоцированы, как правило, на Х-хромосому, реже на одну из аутосом, унаследованных от отца. Транслоцированный участок Y-хромосомы при этом содержит ген SRY. В редких случаях может встречаться скрытый мозаицизм по Y-несущему клону (например, 46,ХY), о чем свидетельствует наличие локусов SRY, AMGL и других Y-специфичных маркеров.
Для идентификации в геноме последовательностей Y-хромосомы, а также выявления делеции гена SRY, используется метод мультиплексной полимеразной цепной реакции (мПЦР), позволяющий исследовать наличия данного гена, а также гена амелогенина (AMG, AMELX), локализованного на коротком плече Х-хромосомы и его гомолога, локализованного на коротком плече Y-хромосомы (AMGL / AMELY). Для анализа наличия точковых мутаций гена SRY – прямое секвенирование его кодирующей последовательности.
С целью анализа количественных аномалий (анеуполидий) и некоторых структурных аномалий половых хромосом, а также гоносомного мозаицизма и химеризма, может быть использован метод количественной флюоресцентной ПЦР.
46,ХХ инверсия пола тип 2 (ОMIM 278850)
Данный тип ХХ-инверсии пола вызван дупликацией регуляторной области гена SOX9. Ген SOX9 располагается на длинном плече хромосомы 17 в локусе q24.3. Кодируемый геном белок является транскрипционным фактором, играющим важную роль в процессе эмбрионального развития. Особенно важен данный белок для развития скелета и репродуктивной системы. Дупликации локуса 17q24 приводит к увеличению числа копий гена SOX9, что у плодов с кариотипом 46,ХХ вызывает дифференцировку гонад по мужскому типу и развитие тестикул в отсутствие гена SRY и других генов Y-хромосомы (SRY-негативная ХХ-инверсия пола). При этом в большинстве случаев дупликаций гена SOX9 не происходит формирования нормальных тестикул, что ведет к овотестикулярной форме нарушения формирования пола (истинный гермафродитизм).
Точковые мутации в гене SOX9 у пациентов с кариотипом 46,ХХ и 46,ХY приводят к развитию кампомелической дисплазии (OMIM 114290) – заболеванию, которое влияет на развитие скелета и репродуктивной системы и часто является угрожающим жизни состоянием в период новорожденности. Пациенты с кариотипом 46,ХХ и точковыми мутациями в гене SOX9 имеют дисгенезию гонад без инверсии пола (фенотипически женщины), а у пациентов с кариотипом 46,XY дисгенезия гонад приводит к XY – инверсии пола или двойственному развитию половых органов.
В Центре Молекулярной Генетики проводится анализ ключевых генов, контролирующих дифференцировку пола: с помощью количественного метода MLPA проводится поиск делеций и дупликаций генов SRY и SOX9.
46,ХХ инверсия пола тип 3 (ОMIM 300833)
Данная форма ХХ-инверсии пола вызвана дупликацией гена SOX3 или делецией его негативно-регуляторной области. Ген SOX3 (sex determining region Y-box 3) является Х-сцепленным гомологом гена SRY, располагается на длинном плече Х-хромосомы в локусе Xq27.1 и подвержен Х-инактивации. Кодируемый им белок является членом семейства SOX (SRY-related HMG-box) – транскрипционных факторов, вовлеченных в контроль дифференцировки различных типов клеток, а также в регуляцию формирования головного мозга в ходе эмбрионального развития, развитие гипоталамо-гипофизарной системы. Он поддерживает недифференцированное состояние у нервных клеток, противодействуя влиянию факторов, стимулирующих их специализацию. Также наличие данного белка необходимо для инициирования формирования пола по мужскому типу. В дифференцирующихся тестикулах белок SOX3 поддерживает дифференцировку и развитие предшественников ‘поддерживающих клеток’ бипотенциальных (индифферентных) гонад в клетки Сертоли, а не в клетки гранулезы овариальной ткани.
Мутации, делеции или дупликации гена SOX3 являются причиной X-сцепленного гипопитуитаризма, некоторых форм аномалии развития нервной трубки (септо-оптической дисплазии), а также X-сцепленной умственной отсталости с изолированным дефицитом гормона роста. Дупликации Xq26-q27, включающие ген SOX3, являются одной из причин SRY-негативной ХХ-инверсии пола (46,ХХ тестикулярной и овотестикулярной форм нарушения формирования пола). Описано несколько случаев ХХ-инверсии пола, вызванных микродупликациями и микроделециями в локусе Xq27.1. При этом спектр фенотипических проявлений (от бесплодия при нормальном мужском фенотипе до аномалий развития пола, микроцефалии с задержкой умственного развития) зависел от локализации и размера перестройки.
Что такое синдром Морриса? Причины возникновения, диагностику и методы лечения разберем в статье доктора Литвинова В. В., репродуктолога со стажем в 38 лет.

Определение болезни. Причины заболевания
Синдром Морриса (синдром тестикулярной феминизации) — это врождённое генетическое заболевание, при котором у людей мужского пола ткани-мишени не чувствительны к мужским половым гормонам — андрогенам. Человек с синдромом Морриса генетически является мужчиной (имеет кариотип 46 XY), но выглядит как женщина.
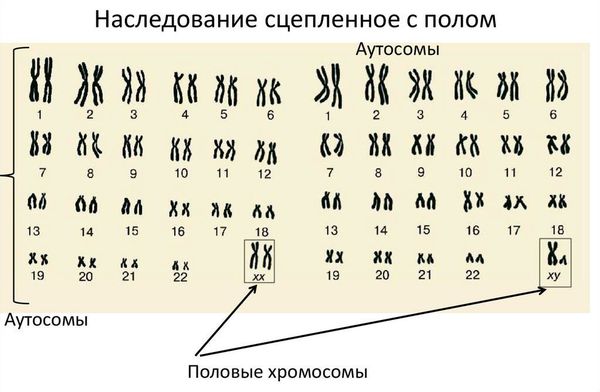
Кариотип — это набор хромосом, который передаётся ребёнку от матери и отца. Кариотип позволяет определить характеристики индивида, включая пол. В норме у человека в генетическом наборе присутствует 46 хромосом, из них 22 пары аутосомных (не определяющих пол) и одна пара половых хромосом, которая определяет гендерную принадлежность ребёнка. Половые хромосомы женщины обозначаются как ХХ, мужчины — ХY. То есть нормальный женский кариотип — 46 XX, мужской — 46 XY.
Часто мужчины с синдромом Морриса даже не догадываются о своём биологическом поле (как и их родители) и живут как девочки/женщины. Это объясняется тем, что данная патология больше никак себя не проявляет, кроме проблем с фертильностью (способностью к зачатию) во взрослом возрасте.


Симптомы синдрома Морриса
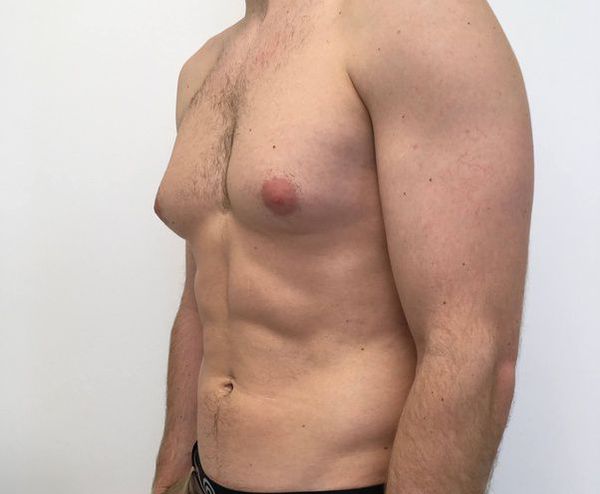
Как правило, люди с синдромом тестикулярной феминизации имеют женский фенотип, т. е. внешне выглядят как женщины. Однако есть некоторые признаки, по которым можно определить наличие заболевания.

Патогенез синдрома Морриса
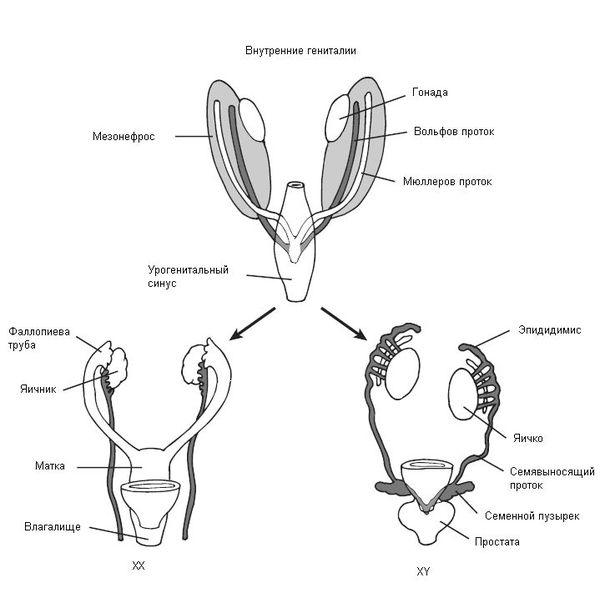
В начале эмбрионального развития у зародышей вне зависимости от хромосомного набора, образовавшегося при оплодотворении яйцеклетки сперматозоидом, половая система закладывается одинаково и предоставляет возможности для развития как женской, так и мужской половой системы. В частности, у зародыша одновременно формируются вольфов и мюллеров протоки, которые потом превращаются в семявыносящие протоки у мужчин и матку с фаллопиевыми трубами и влагалищем у женщин. Половые железы эмбриона (гонады) не дифференцированы и содержат первичные половые клетки (гаметы), которые могут превратиться как в клетки яичников (женские гонады), так и в клетки семенников (мужские гонады).
Таким образом, эмбрион до 6 недель является нейтральным по полу (имеет признаки и мужского, и женского пола). Далее процесс формирования половых признаков и в дальнейшем организма происходит под строгим контролем гормонов: у эмбриона мужского пола — под влиянием тестостерона (влагалище атрофируется), у эмбриона женского пола — под влиянием эстрадиола и прогестерона (влагалище трансформируется из «слепого мешка» и формируется шейка матки и матка). Тестостерон и эстрадиол вырабатываются как у мужчин, так и у женщин, только их соотношение разное. В норме у мальчиков соотношение мужского гормона к женскому 4\1, а в случае СТФ — 0\1. У девочек соотношение мужского гормона к женскому, прямо противоположное — 1\4.
Классификация и стадии развития синдрома Морриса
Синдром тестикулярной феминизации делят на две формы — полный и неполный. Дети с полной формой нечувствительности к мужским гормонам имеют однозначно женский внешний вид. При этом состоянии чувствительность организма к мужскому половому гормону (тестостерону) отсутствует полностью. Рождается здоровая «девочка», не имеющая, на первый взгляд, каких-либо отклонений в развитии.
Осложнения синдрома Морриса
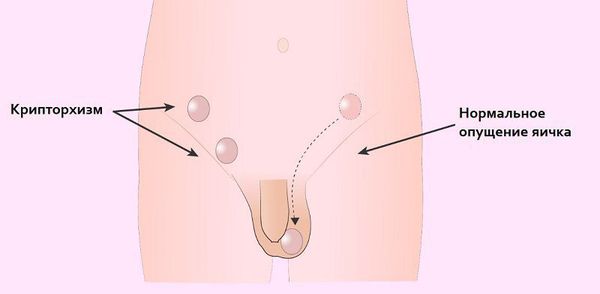
Диагностика синдрома Морриса
Лечение синдрома Морриса
Лечение синдрома тестикулярной феминизации должно осуществляться междисциплинарной командой врачей, которая состоит из хирурга, гинеколога, генетика, эндокринолога и клинического психолога или психиатра.
Операция эффективна в 100 % случаев. Но так как удаляемые гонады мягкие и не имеют чёткой формы и структуры, возможны случаи неполного их удаления. Тогда оставшаяся ткань яичка может возобновить свою работу, что проявляется повышением тестостерона в крови и визуализацией на УЗИ гонады/гонад. В этом случае необходима повторная операция. Однако это случается очень редко.
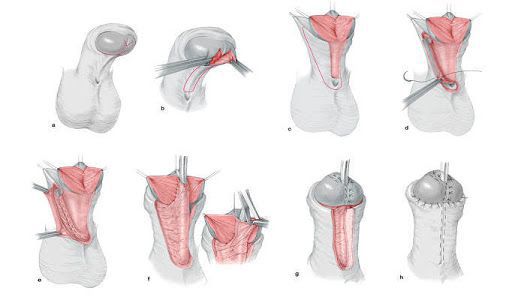
Прогноз. Профилактика
Инверсия пола у людей
Этиология и встречаемость инверсии пола. Инверсия пола — панэтническое и генетически разнородное заболевание. Среди пациентов с полной гонадной дисгенезией наиболее частые причины инверсии пола — точковые мутации, делеции или транслокации гена SRY.
Приблизительно 80% мужчин 46,ХХ с полной дисгенезией гонад имеют транслокацию SRY на Х-хромосому, и 20-30% женщин 46,XY с полной дисгенезией гонад имеют мутацию или делецию гена SRY. Встречаемость мужчин 46,ХХ и женщин 46,XY составляет примерно 1 на 20 000.
Патогенез инверсии пола
SRY — связанный с ДНК белок, изменяющий структуру хроматина, изгибая ДНК. Эта связь и изменение свойств ДНК указывают, что белок SRY регулирует экспрессию генов.
В ходе нормального развития человека SRY необходим для образования мужских половых органов, но для образования женских половых органов его отсутствие не обязательно. Точный механизм влияния SRY на развитие мужских гонад неопределен, хотя некоторые наблюдения указывают, что SRY подавляет отрицательный регулятор развития тестикул.
Мутации в гене SRY, выявляемые у XY-женщин, вызывают потерю его функции. Среди XY-женщин 10-15% имеют делецию SRY (женщины XY, SRY), еще 10-15% имеют точковые мутации в пределах гена. Точковые мутации в гене SRY нарушают связь с ДНК или поворот ДНК.
Нарушение SRY у мужчин XX — транслокация SRY с Yp на Хр (мужчины XX, SRY+; рис. С-36, см. цв. вклейку). В мужском мейозе происходит кроссинговер между псевдоаутосомными регионами Хр и Yp; этот кроссинговер обеспечивает правильное расхождение и соответствие последовательностей в псевдоаутосомных регионах X и Y.
Тем не менее иногда рекомбинация происходит неправильно, захватывая центромеру, что приводит в передаче специфических последовательностей короткого плеча Yp-, включая ген SRY, на короткое плечо Хр.
Кроме SRY, Y-хромосома содержит, по крайней мере, три локуса (локусы факторов азооспермии AZFa, AZFb и AZFc), необходимых для нормального сперматогенеза. Отсутствие этих локусов, по крайней мере, частично, объясняет бесплодие у мужчин XX, SRY+.
Х-хромосома также содержит несколько локусов, необходимых для функционирования яичников и женской фертильности. Для развития овоцитов достаточно наличия одной Х-хромосомы, но для использования овоцитов необходимы две Х-хромосомы. В соответствии с этим наблюдением, у женских плодов XY овоциты закладываются, но фолликулы вырождаются еще до рождения или вскоре после него. Следовательно, бесплодие женщин XY объясняется отсутствием второй Х-хромосомы.
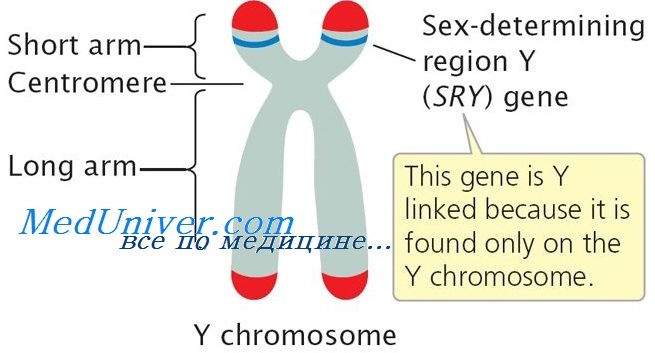
Фенотип и развитие инверсии пола
Мужчины XX, SRY+ имеют много признаков синдрома Кляйнфелтера (47,XXY), включая гипогонадизм, азооспермию, гиалиноз семявыносящих канальцев и гинекомастию. Несмотря на сниженный синтез тестостерона, у большинства пациентов пубертат развивается самостоятельно, хотя для достижения полной вирилизации может потребоваться введение тестостерона.
В отличие от пациентов с синдромом Кляйнфелтера, большинство мужчин 46,ХХ имеют нормальный рост, нормальные пропорции скелета, интеллект и меньше психосоциальных проблем. Пациенты с большим участком Yp в Х-хромосоме имеют большее сходство с синдромом Кляйнфелтера.
Женщины XY, SRY- имеют полную дисгенезию гонад, они обычно выше среднего роста для нормальных женщин. Эти пациентки имеют физические характеристики синдрома Тернера, только если делеция SRY связана с обширной делецией Yp. Поскольку у этих пациенток вместо яичников присутствуют только рубчики (полоски соединительной ткани), спонтанно пубертат у них не развивается.
В отличие от полной пенетрантности и сравнительно однородной экспрессивности, наблюдаемых при транслокациях или делециях гена SRY, точковые мутации SRY проявляют как неполную пенетрантность, так и варьирующую экспрессивность. Пациенты с точковыми мутациями в гене SRY обычно имеют полную дисгенезию гонад, рост выше среднего и у них не развиваются вторичные половые признаки.
Тем не менее описано несколько точковых мутаций SRY, связанных с бесплодием (полной дисгенезией гонад) при женском фенотипе и фертильностью при мужском фенотипе в одной и той же семье.
Особенности фенотипических проявлений инверсии пола:
• Возраст начала: пренатальный
• Бесплодие
• Недоразвитие вторичных половых признаков
• Однозначные гениталии
Лечение инверсии пола
У пациентов с полной дисгенезией гонад диагноз инверсии пола обычно устанавливают при расхождении данных УЗИ плода и кариотипа плода, или из-за отсутствия или неполного развития вторичных половых признаков и бесплодия. Доказательство того, что инверсия пола вторична по отношению к аномалии экспрессии SRY, требует демонстрации соответствующего изменения в гене SRY.
Для мужчин XX, SRY+ назначение андрогенов обычно способствует вирилизации, но лечение, предохраняющее от азооспермии, в настоящее время невозможно. Введение дополнительных андрогенов не помогает избежать гинекомастии. Если гинекомастия становится достаточно выраженной, пациентам приходится прибегать к хирургическому лечению.
Для женщин XY, SRY+ и женщин XY с точковыми мутациями гена SRY терапия эстрогенами обычно начинается в возрасте 14-15 лет, чтобы обеспечить развитие вторичных половых признаков. Для вызывания менструаций в схему лечения добавляют прогестерон либо в момент первого вагинального кровотечения, либо через 2 года после начала лечения эстрогенами.
Кроме того, из-за риска развития опухолей гонад, рекомендуется удаление неполноценных яичников после завершения роста скелета.
Как и при всех нарушениях половой дифференцировки или несоответствиях генетического и фенотипического пола, чрезвычайно важны психосоциальная коррекция и консультирование семьи и пациента. Большинство семей и пациентов имеют затруднения в понимании медицинских данных и получении соответствующих психосоциальных установок.
Риски наследования инверсии пола
Неправильная рекомбинация de novo — наиболее частая причина появления мужчин XX, SRY+ и женщин XY SRY_; следовательно, большинство пар с больным ребенком имеет низкий риск повторения у будущих детей. Тем не менее, иногда мужчины XX SRY+ и женщины XY SRY возникают в результате наследуемой делеции SRY или транслокации от отца со сбалансированной транслокацией между Хр и Yp.
Если отец — носитель транслокации, все дети будут или мальчиками XX, SRY+ или девочками XY, SRY-. Поскольку мужчины XX, SRY+ и женщины XY,SRY всегда бесплодны, они не могут передать заболевание потомкам.
Большинство женщин XY с точковыми мутациями в гене SRY имеют новые мутации. Родители больного ребенка, следовательно, обычно имеют низкий риск повторения у будущих детей; тем не менее, поскольку некоторые мутации SRY имеют неполную пенетрантность, здоровые фертильные отцы могут нести мутации SRY, способные вызвать инверсию пола у их детей с генотипом XY.
Пример инверсии пола. Женщина, 37-летняя служащая, беременна первым ребенком. Из-за возрастного риска иметь ребенка с хромосомной аномалией она решила провести амниоцентез, чтобы оценить кариотип плода; результат кариотипа нормальный, 46.ХХ. Тем не менее на 18-й нед гестации при УЗИ обнаружен нормальный плод мужского пола; последующее подробное УЗИ подтвердило мужской пол плода.
Женщина была здорова перед и в течение беременности, без влияния инфекций или лекAPCтв в ходе беременности. Ни она, ни ее партнер не имели в семейном анамнезе случаев неоднозначных половых органов, бесплодия или врожденных пороков.
Повторный хромосомный анализ подтвердил нормальный кариотип 46.ХХ, но методом FISH выявлено присутствие сигнала гена региона половой детерминации Y (SRY) в одной из Х-хромосом. На 38-й нед беременности пациентка без осложнений самостоятельно родила фенотипически нормального ребенка мужского пола.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021



