Симптомы, причины и методы лечения кольцефалии
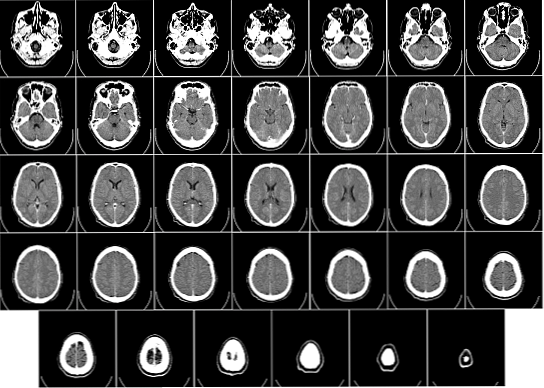
colpocephaly это врожденная аномалия головного мозга, которая влияет на структуру желудочковой системы (Esenwa & Leaf, 2013). На анатомическом уровне в головном мозге наблюдается значительное расширение затылочных рогов боковых желудочков (Pérez-Castrillón et al., 2001).
Можно наблюдать, как задняя часть боковых желудочков больше, чем ожидалось, из-за аномального развития белого вещества (Национальный институт неврологических расстройств и инсульта, 2015).
Клиническое проявление этой патологии проявляется в первые годы жизни и характеризуется когнитивной, двигательной задержкой созревания и развитием судорожных эпизодов и симптомов эпилепсии (Bartolomé et al., 2013).
Хотя конкретная причина colpocephaly до сих пор не обнаружена, этот тип патологии может быть результат развития какого-то ненормального процесса во время эмбрионального развития во второй и шестую месяцы беременности (Национальный институт неврологических расстройств и инсульта, 2015 ).
Диагноз кольпоцефалия обычно ставится в до или перинатальном периоде (Esenwa & Leaf, 2013), диагноз во взрослой жизни очень редок (Bartolomé et al., 2013).
С другой стороны, прогноз людей, страдающих кольцецефалией, зависит главным образом от тяжести патологии, степени развития мозга и наличия других видов медицинских осложнений (Национальный институт неврологических расстройств и инсульта, 2015).
Медицинское вмешательство при кольцецефалии в основном ориентировано на лечение вторичных патологий, таких как судороги (Национальный институт неврологических расстройств и инсульт, 2015).
Характеристика кольцефалии
colpocephaly является врожденным неврологическим расстройством, то есть происходит изменение нормального и эффективного развития нервной системы, в данном случае, различных областей мозга во время беременности.
В частности, врожденные изменения, затрагивающие центральную нервную систему (ЦНС), являются одной из основных причин смертности и заболеваемости плода (Piro, Alongi et al., 2013).
Всемирная организация здравоохранения (ВОЗ) отмечает, что около 276 000 новорожденных умирают в течение первых четырех недель жизни в результате некоторых врожденных патологий..
Кроме того, этот тип аномалий представляет собой одну из наиболее важных причин нарушения функциональности в детской популяции, так как они вызывают широкий спектр неврологических расстройств (Herman-Sucharska et al, 2009).
С другой стороны, кольцецефалия классифицируется в группе патологий, которые влияют на структуру мозга и известны как расстройства мозга«.
расстройства мозга они относятся к наличию различных изменений или аномалий центральной нервной системы, возникших на первых этапах развития плода (Quenta Huayhua, 2014).
Развивается нервная система (SN), в дородовой и послеродовой следует ряд процессов и событий высокой сложности, главным образом, на основе различных нейрохимических событий, генетически запрограммированы и на самом деле подвержены воздействию внешних факторов, таких как влияние окружающей среды.
Когда это происходит врожденный порок развития нервной системы, начать развивать структуры и / или функции ненормальным образом, что имеет серьезные последствия для человека, как физически, так и когнитивно.
В частности, кольцецефалия является патологией, которая влияет на развитие боковые желудочки, в частности, в задние или затылочные области (Gary et al., 1992), что приводит к аномально большому росту затылочных канавок.
Этот врожденный порок развития, известный как кольцецефалия, был первоначально описан Бенда в 1940 году и упоминается в различных клинических докладах Яковлев и Уодсуорт в 1946 году (Нигро и др., 1991).
Однако позже этот термин colpocephaly был использован для обозначения расстройства, характеризующегося персистирующей фетальной конфигурацией мозга желудочков, где затылочные рога представлены дилатационными и аномально большими (Flores-Sarnat, 2016).
Хотя это медицинское состояние мало рассмотрено в медицинской и экспериментальной литературе, оно было связано с наличием судорожные приступы, умственная отсталость и различные сенсорные и моторные изменения (Нигро и др., 1991).
статистика
Кольцефалия является очень редким врожденным пороком развития. Хотя в последнее время нет данных, по состоянию на 1992 год было описано приблизительно 36 различных случаев людей, пораженных этой патологией (Gary et al., 1992)
Отсутствие статистических данных об этой патологии может быть результатом отсутствия консенсуса по клиническим характеристикам, а также из-за диагностических ошибок, так как это связано с различными заболеваниями..
Признаки и симптомы
Характерным структурным признаком кольцецефалии является наличие расширения или уплощения затылочных рогов боковых желудочков (Nigro et al., 1991)..
Внутри нашего мозга мы можем найти систему полостей, сообщающихся друг с другом и омываемых спинномозговой жидкостью (CSF), желудочковой системой (Waxman, 2011).
Эта жидкость содержит белки, электролиты и некоторые клетки. Помимо защиты от возможной травмы, спинномозговая жидкость играет важную роль в поддержании церебрального гомеостаза благодаря своей питательной, иммунологической и воспалительной функции (Chauvet and Boch, X).
боковые желудочки они являются самыми большими частями этой желудочковой системы и образованы двумя центральными областями (тело и предсердие) и тремя расширениями (рога) (Waxman, 2011)
В частности, задний рог или затылок, он простирается до затылочной доли, а его крыша образована различными волокнами мозолистого тела (Waxman, 2011).
Следовательно, любой тип изменения, который вызывает порок развития или различные травмы и повреждения в боковых желудочках, может привести к широкому разнообразию неврологических признаков и симптомов..
В случае colpocephaly, наиболее распространенные клинические признаки включают в себя: детский церебральный паралич, интеллектуальный дефицит, микроцефалия, Миеломенингоцеле, агенезия мозолистого тела, lisecefalia, гипоплазии мозжечка, моторные нарушения, мышечные спазмы, судорожные эпизоды и гипоплазия зрительного нерва (Gary ЕТ аль, 1992 ;. Квэнта Huayhua, 2014).
микроцефалия
Это редкое или редкое изменение, однако степень выраженности микроцефалии варьируется, и многие дети с микроцефалией могут испытывать различные изменения, а также неврологические и когнитивные задержки (Бостонская детская больница, 2016 г.).
Возможно, что у тех людей, у которых развивается микроцефалия, возникают рецидивирующие судорожные приступы, различные физические нарушения, дефицит обучения, среди прочего (World Health Organization, 2016).
Церебральный паралич
Термин церебральный паралич (ПК) относится к группе неврологических расстройств, которые затрагивают области, отвечающие за моторный контроль (Национальный институт неврологических расстройств и инсульта, 2016).
Повреждения и травмы обычно имеют место во время развития плода или в начальные моменты постнатальной жизни и будут постоянно влиять на движения тела и координацию мышц, но не будут постепенно увеличивать тяжесть (Национальный институт Неврологические расстройства и инсульт, 2016).
Обычно церебральный паралич вызывает физическую инвалидность, которая варьируется по степени вовлеченности, но также может сопровождаться сенсорной и / или интеллектуальной инвалидностью (ASPACE Confederation, 2012).
Следовательно, различные сенсорные, когнитивные, коммуникационные, воспринимаемые, поведенческие, эпилептические кризисы и т. Д. Дефициты могут появляться в связи с этой патологией. (Muriel et al., 2014).
миеломенингоцеле
Со сроком mielomeningocele мы ссылаемся на один из типов расщелина позвоночника.
Спина позвоночника является врожденный дефект, который влияет на различные структуры спинного мозга и позвоночника и в дополнение к другим изменениям, может вызвать паралич конечностей или нижних конечностей (World Health Organization, 2012).
Исходя из пораженных участков, мы можем дифференцировать четыре типа расщелины позвоночника: оккультные, дефекты закрытой нервной трубки, менингоцеле и миеломенингоцеле (NationalInstitu of NeurologicalDisorders and Stroke, 2006).
В частности, миеломенигоцеле, также известное как открытая расщелина позвоночника, считается наиболее тяжелым подтипом (Mayo Clinic, 2014)..
На анатомическом уровне можно наблюдать, как позвоночный канал открыт или открыт вдоль одного или нескольких позвоночных сегментов, в средней или нижней части спины. Таким образом, мозговые оболочки и спинной мозг выступают в виде мешочка на спине (Mayo Clinic, 2014).
Как следствие, люди с диагнозом миеломенингоцеле могут проявлять значительное неврологическое поражение, которое включает такие симптомы, как: мышечная слабость и / или паралич нижних конечностей; кишечные изменения, судороги и ортопедические изменения, среди прочего (Клиника Майо, 2014).
Агенезия мозолистого тела
агенезия мозолистого тела представляет собой тип неврологической патологии Врожденные относится к частичному или полному отсутствию структуры, которая соединяет полушария головных мозга, мозолистое тело (Национальный институт неврологических disordes и инсульт, 2014).
Этот тип патологии обычно связан с другими заболеваниями, такими как порок развития Киари, синдром Ангельмана, синдром Денди-Уокера, шикефалия, голопрозэнцефалия и т. Д. (Национальный институт неврологических расстройств и инсульта, 2014).
Клинические последствия агенеза мозолистого тела значительно различаются среди пораженных, хотя некоторые общие характеристики: дефицит взаимосвязи зрительных образов, умственная отсталость, судороги или спастичность (Национальный институт неврологических расстройств и инсульт, 2014).
Лиссэнцефалия
Lissencephaly является врожденным пороком развития, который также является частью группы энцефальных расстройств (Национальная организация по редким расстройствам, 2012).
Эта патология характеризуется отсутствием или частичным развитием мозговых извилин коры головного мозга (Национальная организация по редким заболеваниям, 2012)..
Поверхность головного мозга имеет необычно гладкий вид и может привести к развитию микроцефалии, изменениям лица, психомоторной отсталости, мышечным спазмам, судорогам и т. Д. (Национальный институт неврологических расстройств и инсульта, 2010).
конвульсии
Судорожные эпизоды или эпилептические припадки возникают в результате необычной нейронной активности, то есть привычная деятельность изменяется, вызывая припадки или периоды необычного поведения и ощущений, и иногда может привести к потере сознания (Mayo Clinic., 2015 ).
Симптомы судорог и эпилептических припадков могут значительно различаться в зависимости от области мозга, в которой он возникает, и от человека, который страдает (Mayo Clinic., 2015).
Некоторые из клинических характеристик судорог: временное замешательство, неконтролируемое дрожание конечностей, потеря сознания и / или отсутствие эпилепсии (Mayo Clinic., 2015).
Эпизоды в дополнение к постановке ситуации, представляющей опасность для человека, подверженного риску падений, утоплений, дорожно-транспортных происшествий и т. Д., Являются важным фактором в развитии повреждения головного мозга из-за ненормальной активности нейронов..
Гипоплазия мозжечка
гипоплазия мозжечка неврологическая патология, характеризующаяся отсутствием полного и функционального развития мозжечка (гипоплазия, 2013).
В последнее время различные исследования показали тесную связь между структурными и функциональными аномалиями мозжечка и различными психическими расстройствами, особенно шизофренией (Chen et al., 2013; Fatemi et al., 2013), биполярным расстройством (Baldacara et al., 2011; Liang et al., 2013), депрессия, тревожные расстройства (Nakao et al., 2011; Schutter et al., 2012; Talati et al., 2013), синдром дефицита внимания с гиперактивностью (ADHD) (An et al. al., 2013; Tomasi et al., 2012; Wang et al., 2013) и аутизм (Marko et al., 2015; Weigiel et al., 2014).
Гипоплазия зрительного нерва
гипоплазия зрительного нерва это другой тип неврологического расстройства, которое влияет на развитие зрительных нервов. В частности, зрительные нервы меньше, чем ожидалось, для пола и возрастной группы больного человека (Hypoplasia, 2013).
Среди медицинских последствий, которые могут возникнуть в результате этой патологии, мы можем выделить: снижение зрения, частичную или полную слепоту и / или аномальные движения глаз (Hypoplasia, 2013).
Помимо зрительных изменений, гипоплазия зрительного нерва обычно связана с другими вторичными осложнениями, такими как: когнитивный дефицит, синдром Морсье, моторные и языковые расстройства, гормональный дефицит и др. (Hypoplasia, 2013).
Интеллектуальный дефицит и моторные изменения
В результате кольпоцефалического покрытия пострадавшие могут проявлять генерализованную когнитивную задержку созревания, то есть развитие их навыков внимания, лингвистики, памяти и практики будет ниже, чем ожидалось для их возрастной группы и уровня образования..
С другой стороны, среди изменений, связанных с двигательной сферой, могут быть мышечные спазмы, изменение мышечного тонуса, среди других симптомов.
причины
Кольпоцефалия возникает при недостаточном утолщении или миелинизации затылочных областей (Quenta Huayhua, 2014).
Хотя причина этого изменения неизвестна, генетические мутации, нарушения миграции нейронов, облучение и / или потребление токсических веществ или инфекций, как возможные этиологические причины кольцефалия (Quenta Huayhua, 2014).
диагностика
Кольцецефалия является типом пороков развития головного мозга, который можно диагностировать до рождения, если можно продемонстрировать наличие увеличения затылочных рогов боковых желудочков (Gary et al., 1992).
Некоторые из диагностических методов, используемых в этой патологии: ультразвук ультразвуком, магнитный резонанс, компьютерная томография, пневмоэнцефалография и вентрикулография.
Есть ли лечение от кольцецефалии?
В настоящее время нет специфического лечения кольцецефалии. Следовательно, вмешательства будут зависеть от степени поражения и от симптомов, вторичных к этой патологии..
Как правило, вмешательства направлены на контроль эпизодов судорог, профилактику мышечных расстройств, восстановление двигательной функции и когнитивную реабилитацию (Gary et al., 1992)..
Агенезия мозолистого тела
Агенезия мозолистого тела — это врожденное отсутствие мозолистого тела либо его части. Аномалия обусловлена генетическими нарушениями, сосудистыми мальформациями, тератогенными факторами. Основные признаки заболевания: двигательные расстройства, задержка психоречевого развития, судорожные приступы. При негрубом (частичном) варианте патологии возможно малосимптомное течение. Для диагностики состояния назначается церебральные КТ или МРТ, нейросонография у новорожденных, генетические исследования. Лечение симптоматическое: медикаментозная коррекция осложнений, реабилитационные программы.
МКБ-10
Общие сведения
Агенезия мозолистого тела (АМТ) — один из наиболее частых пороков нервной системы. Распространенность болезни в популяции составляет от 0,05% до 7% среди новорожденных, причем в группе детей с замедленным становлением психики агенезия встречается у 2,3%. Калифорнийская программа по изучению врожденных пороков предоставляет другие данные по частоте агенезии — 1,4 на 10000 живых новорожденных. Впервые состояние было описано в 1812 году в ходе аутопсии, проведенной немецким анатомом И. Рэйлем, и названо «природной моделью рассеченного мозга».
Причины
Точные этиологические факторы заболевания не установлены. В современной неврологии преобладает мультифакториальная теория, согласно которой для формирования врожденного порока ЦНС требуется комбинация неблагоприятных экзогенных и эндогенных причин. Ученые выделяют несколько наиболее вероятных предпосылок развития агенезии:
Основным фактором риска выступает недоношенность. У новорожденных, родившихся до 27-недельного срока гестации МТ истончено в задних отделах, между 28 и 30 неделями — только в области валика. У рожденных после 30 недели в неонатальном периоде изменения не обнаруживаются, хотя при нейропсихологическом исследовании у школьников зачастую выявляется дефицит межполушарной передачи познавательной информации.
Патогенез
Мозолистое тело (МТ) представляет собой крупный пучок комиссуральных нервных волокон. Это важный аксональный путь, который соединяет соответствующие зоны коры правого и левого полушарий. Анатомическая структура имеет длину 7-9 см, состоит из более 300 млн. аксонов. Формирование МТ начинается на этапе позднего нейроонтогенеза (8-9 недели эмбриогенеза). Его созревание в норме продолжается до 20-25 лет.
Агенезия возникает при нарушении дифференциации нервной трубки в период со 2 до 5 месяца внутриутробного развития. При полном отсутствии МТ третий мозговой желудочек остается открытым, не формируются столбы свода мозга, отсутствуют прозрачные перегородки. В 60% случаев при АМТ передней комиссуры нет вообще. В 10% она увеличена и берет на себя часть функций мозолистого тела у новорожденных, а также на следующих этапах постнатального периода.
Характерным анатомическим изменением является колпоцефалия, при которой расширены задние отделы боковых церебральных желудочков. Состояние не относится к истинной гидроцефалии новорожденных, а обусловлено уменьшением кортикальных ассоциативных путей. Еще один типичный признак порока — пучки Пробста, представляющие собой неправильно ориентированные аксоны, расположенные параллельно межполушарной щели.
Классификация
В практической неврологии состояние подразделяют на тотальное, когда орган полностью отсутствует, и частичное (парциальное), при котором визуализационные методы не обнаруживают отдельные участки МТ. Это имеет решающее значение для тяжести клинической картины, возможных осложнений. В соответствии с патогенетическими особенностями формирования врожденных пороков, выделяют следующие 3 формы болезни:
Симптомы
Клиническая картина агенезии мозолистого тела широко варьирует от практически бессимптомных форм (при гипоплазии) до критических нервно-психических расстройств при его грубом недоразвитии, сопровождающемся другими врожденными пороками ЦНС. У новорожденных признаки патологии могут вовсе отсутствовать и проявляться по мере взросления младенца задержкой психомоторного развития.
Двигательные нарушения определяются у 35-40% пациентов. Они проявляются мышечной гипотонией или дистонией, гипер- или гипорефлексией, нарушением глотательного и сосательного рефлексов. Дети позже начинают держать голову, испытывают затруднения при обучении сидению, ползанию, ходьбе. Могут отмечаться координационные нарушения, неуклюжая походка. Из пароксизмальных расстройств у новорожденных и детей первого года жизни преобладают судороги.
Мозолистое тело поддерживает связь между церебральными зонами, формирует межполушарную организацию высших психических процессов. При его агенезии либо гипоплазии у детей выявляются когнитивные расстройства. У новорожденных пациентов и в раннем детстве наблюдается задержка речи, снижение динамического компонента игровой деятельности. В дошкольном и школьном возрасте возникают проблемы с концентрацией внимания, расстройства памяти, при тотальной АМТ снижен коэффициент интеллекта.
Осложнения
Около 65% случаев заболевания сопровождаются сопутствующими врожденными патологиями, среди которых преобладают мальформации кортикального развития (22,8%), межполушарные кисты (14,3%), голопрозэнцефалия (14,3%). К более редким сопутствующим аномалиям относят кисты и гипоплазию мозжечка, синдром Арнольда-Киари. До 20% новорожденных, кроме структур ЦНС, имеют пороки нескольких внутренних органов.
У 75% больных с тотальным поражением наблюдается симптоматическая эпилепсия височно-лобной локализации, в 66% случаев выражены когнитивные нарушения. У 16% пациентов формируются расстройства аутистического спектра. Изредка встречаются патологии органа зрения в виде хориоретинальных лакунарных очагов, сочетанной аномалии зрительных нервов.
Диагностика
В качестве первичного метода обследования в пренатальном периоде проводится акушерское УЗИ. У новорожденных для скрининговой диагностики используется нейросонография, однако этот метод не всегда показывает хорошую информативность, особенно при парциальной агенезии. Для верификации диагноза назначаются следующие методы исследования:
Лечение агенезии мозолистого тела
Специфическая терапия отсутствует. Медикаментозное лечение назначается неонатологом или педиатром индивидуально с учетом ведущих патологических синдромов: у новорожденных, детей раннего возраста используются антиконвульсанты, нейрометаболические препараты, дегидратационная терапия. Основу медицинской помощи составляет комплексная реабилитация, которая включает следующие составляющие:
Прогноз и профилактика
Прогноз определяется видом врожденной аномалии мозолистого тела, наличием сопутствующих пороков развития ЦНС. Благоприятный исход наблюдается при частичной гипоплазии МТ, а в случае комбинированных церебральных пороков у новорожденных могут быть жизнеугрожающие осложнения. Профилактические меры включают медико-генетическое консультирование, исключение тератогенных влияний в гестационном периоде.
Нейросонография: врожденные аномалии
АВТОР: Lori L. Barr
Ключевые слова: Нейросограмма, нейросонография, ультразвуковое исследование головы
Из-за низкой стоимости, мобильности и безопасности, нейросонография остается предпочтительным методом ранней визуализации головного мозга в период, пока роднички открыты. Эта статья представляет основные сведения для улучшения диагностики неврологической патологии, которая развивается у младенцев. Этот навык лучше всего осваивается путем фактического сканирования пациентов после первоначального скрининга высококвалифицированным специалистом ультразвуковой диагностики. Во всех случаях, будь то это врожденная, инфекционная, неопластическая или травматическая патология, нейросонография – зачастую является первым диагностическим шагом. Дополнительные методы посрезовой визуализации используются для оценки структур и функций, которые с трудом визуализируются с помощью нейросонографии, а именно: субарахноидальное пространство при кровоизлияниях; оценка опухолей или пространство-занимающих поражений в рамках предоперационного планирования; когда стоит вопрос об уточнении процесса демиелинизации.
Современные достижения в нейросонографии, такие как трехмерная (3D) визуализация, количественная характеристика тканей, использование контрастного усиления все еще находятся на стадии исследования клинического внедрения. Продолжение исследований в этих специализированных областях будет способствовать более точному измерению значимых параметров, которые важны для прогнозирования результатов лечения пациентов.
ВРОЖДЕННЫЕ АНОМАЛИИ
Разделение врожденных пороков развития на 4 подгруппы на основе сроков эмбриологического неврологического развития, предложенное Ван дер Кнаппом и Валком, остается принятым стандартом для деления врожденных пороков развития на категории. Среди 4 подгрупп выделяют: дорзальная индукция (первичная нейруляция на 3-4-й неделе гестационного возраста [ГВ], вторичная нейруляция, на 4-40-й неделе ГВ); вентральная индукция (на 5-8-й неделе ГВ); нейронная пролиферации, дифференцировка и гистогенез (на 8-16-й неделе ГВ); миграция нейронов (на 8-20-й неделе ГВ через 1 год после рождения). В статье обсуждаются аномалии, при которых ультразвуковое исследование помогает поставить диагноз.
Пре- и постнатальная визуализация важны из-за хрупкости оболочек над обнаженными элементами центральной нервной системы (ЦНС) у пациентов с цефалоцеле, менингоцеле, миеломенингоцеле и другими дефектами закрытия (рис.1).

Рис. 1. Цефалоцеле. (A) Левая фронтальная сагиттальная нейросограмма у новорожденного с большим мягкотканным новообразованием волосистой части кожи головы демонстрирует энцефалоцеле, которое содержит как цереброспинальную жидкость (C), так и часть лобной доли (F). (B) Коронарная трансабдоминальная нейросограмма плода через заднюю часть головы демонстрирует большое затылочное энцефалоцеле, содержащее большую часть мозжечка (C). Обратите внимание на отсутствие эхогенного свода черепа вокруг мозжечка.
Пренатальное сканирование плодов между 11-й и 13-й неделями ГВ должно позволять визуализировать четвертый желудочек в качестве измеримой внутричерепной прозрачности в срединно-сагиттальной проекции, которая сейчас популярна для измерения затылочной прозрачности. Если четвертый желудочек не виден, это указывает на дефект нервной трубки. Магнитно-резонансная томография (МРТ) – метод выбора после рождения. При визуализации истинного цефалоцеле, менингоцеле или миеломенингоцеле преследуется две цели, а именно идентификация степени дефекта нервной трубки и наличие содержимого мешка. В частности, вопрос заключается в том, есть ли в мешке нервные элементы, поскольку хирургическое вмешательство при этом изменяется.
Порок развития Киари
Нейросонография эффективна при идентификации, проведении хирургического вмешательства и последующем динамическом наблюдении пороков развития Киари. Младенцы с Киари I могут существовать с признаками патологии, которые являются бессимптомными и обнаруживаются случайно (рис.2).

Рис. 2. Порок развития Киари I. Сагиттальная нейросограмма демонстрирует отсутствие жидкости в большой цистерне (стрелка). Обратите внимание, что четвертый желудочек смещен каудально и несколько сплющен (изогнутая стрелка).
Если у этих пациентов появляются патологические признаки, интраоперационная нейросонография может сыграть важную роль при анализе процесса. Как правило, хирург хочет знать, как далеко вниз по цервикальному каналу пролабируют миндалины с пульсацией цереброспинальной жидкости (ЦСЖ), чтобы свести к минимуму развитие сиринкса. Младенцы с Киари II, дисгенезом заднего мозга, обычно представлены при рождении в сочетании с расщелиной позвоночника (spina bifida). Нейросонография эффективна для диагностики вторичной гидроцефалии в динамике после закрытия дефекта нервной трубки. Сонографические находки при Киари II включают смещение вниз и увеличение четвертого желудочка, выступающее срединное тело, облитерацию большой цистерны, низкая направленность передних рогов боковых желудочков и вдавление серпа мозга (рис.3).

Рис. 3. Мальформация Киари II. (A) Сагиттальная нейросограмма новорожденного демонстрирует нисходящее смещение и расширение четвертого желудочка (4) и выпуклость срединного тела (M). Большая цистерна облитерирована. (B) Заднее коронарное изображение через головной мозг и мозжечок демонстрирует выпячивание бокового (L) и третьего (3) желудочков с аномально сформированным четвертым желудочком (стрелка). (C) Переднее коронарное изображение через голову младенца демонстрирует низкую направленность передних рогов (A) боковых желудочков и вдавление серпа мозга (стрелка).
Другими характерными находками являются кольпоцефалия, гидроцефалия, дисгенезия мозолистого тела и сирингогидромиелия. Серийные нейросограммы выполняются с одинаковой глубиной изображения, поэтому размер желудочков легко сравнивается, если не используется 3D-изображение. Гидроцефалия в этих случаях обычно вызывается стенозом водопровода (рис. 4).

Рис. 4. Стеноз водопровода. (A) Сагиттальная нейросограмма показывает увеличение третьего (3) и бокового (L) желудочков. Обратите внимание на пролабирование мозжечковых миндалин в отверстие (стрелка). (B) Коронарная нейросограмма показывает увеличенный боковой (L) и третий (3) желудочки.
Вентральная индукция ведет к образованию заднего мозга, среднего мозга, переднего мозга и лица. Аномалии вентральной индукции включают аномалии гипоталамо-гипофизарной оси, пороки развития мозжечка, дорзальные кисты, голопрозэнцефалию и агенезию/дисгенезию прозрачной пластинки. Отклонения гипоталамо-гипофизарной оси очень трудно диагностировать с помощью ультразвука, в то время как другие категории обнаруживаются только на нейросограммах, если при этом оператор знаком с возможным диагнозом и характерными признаками. Когда нейросонография выполняется с набором этих знаний, МРТ может быть отложена до более позднего младенческого периода, когда становится более важной клиническая одновременная оценка прогресса миелинизации.
Мозжечковые аномалии – это нарушения вентральной индукции. Пренатальный 3D-ультразвуковое исследование очень перспективно в отношении точной диагностики этих аномалий. Кистозные пороки развития задней ямки являются результатом дисгенезии палеоцеребеллума (клочка и червя) и являются общепринятыми. Эти пороки развития включают спектр Денди-Уокера и мегалию большой цистерны. Изолированные аномалии червя и синдром Жубера также являются палеоцеребеллярными по происхождению. Новый мозжечок (неоцеребеллюм) состоит из остальной части полушарий мозжечка. Дисгенезия нового мозжечка приводит к комбинированной мозжечковой гипоплазии, мозжечковой полусферической аплазии/гипоплазии и дисплазии мозжечка.
Спектр Данди-Уокера включает в себя порок развития Дэнди-Уокера, вариант Денди-Уокера и мегалию большой цистерны. Наиболее тяжелая форма, порок развития Дэнди-Уокера, состоит из кистозной дилатации четвертого желудочка, восходящего смещения мозжечкового намета, приводящего к увеличению задней ямке и агенезии червя (рис.5).

Рис. 5. Порок развития Дэнди-Уокера. (A) Задняя коронарная нейросограмма демонстрирует увеличенную заднюю ямку и кистозную дилатацию четвертого желудочка (4). Мозжечковый червь отсутствует. (В) Правая парасагиттальная нейросограмма показывает приподнятый намет мозжечка (стрелка), большой четвертый желудочек (4) и часть правого полушария мозжечка (С). Мозжечковый червь более эхогенный, чем полушария.
Эти находки обычно осложняются гидроцефалией с течением времени. Сопутствующие церебральные аномалии развиваются в 68% случаев. Вариант характеризует случаи, которые не демонстрируют классические признаки порока развития Денди-Уокера. Мегалия большой цистерны – это увеличение большой цистерны без сопутствующих аномалий, которая считается благоприятным вариантом с хорошим долгосрочным прогнозом. Очень плохой прогноз при синдроме Данди-Уолкера и варианте Данди-Уолкера, главным образом за счет связанных с ними аномалий.
Мозжечковая гипоплазия и дисплазия. Гипоплазия и дисплазия червя.
Новая классификация мозжечковых аномалий была предложена Пателем и Барковичем в 2003 году. Дифференциальная диагностика гипоплазии от дисплазии и диффузного и фокального заболевания важна для выделения младенцев с сопутствующими церебральными аномалиями и детей с изолированными мозжечковыми аномалиями. Наличие сопутствующих аномалий связано с плохим прогнозом. Постнатальная МРТ-визуализация является золотым стандартом для классифицирования, при этом дифференциальная диагностика с помощью ультразвукового исследования гипоплазии от дисплазии не является достоверной. Агенезия определяется при отсутствии эхогенного червя и полушарий мозжечка. Гипоплазия червя характеризуется сглаживанием нижней части червя в средне-сагиттальной проекции. Асимметрия полушарий мозжечка или небольшой размер является ключом к диагностике гипоплазии мозжечка (рис. 6).

Рис. 6. Гипоплазия мозжечка и червя. Коронарная нейросограмма демонстрирует небольшие асимметричные полушария мозжечка (C) и отсутствие червя. У этого пациента также имеются признаки порока развития Дэнди-Уокера и отсутствии мозолистого тела.
Три синдрома связаны с полной агенезией червя: мозжечково-глазной мышечный синдром, синдром Жубера и синдром Уокера-Варбурга. Агенезия червя может присутствовать при синдроме Коффина-Сириса, синдроме криптофтальмии, синдроме Эллиса ван Кревельда и синдроме Меккеля-Грубера.
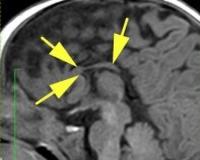
Аплазии/гипоплазии мозолистого тела
Это расстройство вентральной индукции является результатом нарушения расщепления переднего мозгового пузыря. Аплазия связана с судорогами и умственной отсталостью. Нейросонографические данные зависят от части (клюв, колено, тело и валик) мозолистого тела, которая отсутствует или истончена (рис.7).

Рис. 7. Аплазия мозолистого тела. (A) Коронарная нейросограмма показывает параллельную ориентацию боковых желудочков. (B) Сагиттальная проекция демонстрирует радиальный порядок борозд и отсутствие мозолистого тела.
Изолированная агенезия имеет хороший долгосрочный прогноз развитии нервной системы в 80% случаев. Гипоплазия также достаточно распространена, при этом оба эти состояния связаны с преждевременным родами, внутриутробной инфекцией и большим возрастом матери.
Голопрозенцефалия – это расстройство вентральной индукции с неполным расщеплением переднего мозгового пузыря. Существует сильная связь дефектов средней линии, которые вовлекают лицо и тело, с факторами окружающей среды и 7 генами, которые рассматриваются в качестве причин развития. Пренатальная диагностика основывается на ультразвуковом исследовании и МРТ. Многие родители предпочитают прерывание беременности. Послеродовая нейросонография и МРТ помогают мультидисциплинарной группе корригировать проблемы, связанные с пациентом.
Наиболее тяжелой формой является алобарная голопрозенцефалия. Мозг состоит только из плоского слоя сросшихся спереди мозговых полушарий в лобной части и одного желудочка, который сообщается с большой дорзальной кистой. Серп мозга отсутствует, а таламусы срощены между собой. Рельефные признаки довольно редкие с гладкой формой на поверхности мозга. Часто встречаются аномалии миграции. Сосудистые аномалии включают отсутствие или одиночные внутренние мозговые артерии, отсутствие мозговой вены, верхнего синуса, сагиттального синуса и прямого синуса. Эта форма голопрозэнцефалии связана с трисомией 13 и с трисомией 18.
Менее выраженная форма представляет собой полулобарную голопрозэнцефалию, где межполушарная щель развивается сзади, но при этом неполная спереди. Частичное слияние таламуса вдоль дна недоразвитого третьего желудочка, что является отличительным признаком (рис.8). Развивается только один желудочек. Отдельные части прозрачной перегородки и мозолистого тела могут полностью отсутствовать.

Рис. 8. Полулобарная голозпроэнцефалия. Коронарная нейросограмма демонстрирует слияние таламусов (Т) и один желудочек.
Самая легкая форма голопрозэнцефалии – лобарная. При этом имеет место неполная межполушарная борозда в передне-задней части и развивается вдавление серпа мозга. Кора головного мозга срощена в зоне лобного полюса с отсутствующей прозрачной перегородкой. Мозолистое тело обычно недоразвитое. Связанные аномалии миграции, такие как гетеротопия серого вещества, с легкостью оцениваются с помощью высокочастотных датчиков в диапазоне частот от 10 до 13 МГц.
Агенезия и дисгенезия прозрачной перегородки
Хотя и встречается первичная агенезия прозрачной перегородки, более распространенными являются вторичная деструкция. Эта деструкция может развиваться после травмы, воспаления или обструкции желудочков. Коронарная проекция является наиболее полезной для демонстрации отсутствия прозрачной перегородки (рис. 9).

Рис. 9. Отсутствие прозрачной перегородки. (A) Коронарная нейросограмма, полученная через передний родничок, демонстрирует отсутствие прозрачной перегородки и квадратную форму передних рогов боковых желудочков. (B) Коронарная нейросограмма демонстрирует нормальную прозрачную перегородку (стрелки) и разделенную полостью прозрачную перегородку для сравнения.
Многие аномалии связаны с отсутствием прозрачной перегородки, включая стеноз водопровода со вторичной гидроцефалией, агенезию мозолистого тела, порок развития Киари II, аномалии миграции и септо-оптическую дисплазию. Септо-оптическая дисплазия включает в себя отсутствие прозрачной перегородки с гипоплазией зрительных каналов, хиазмы и нервов. Большинство людей с септо-оптической дисплазией также имеют нарушения гипоталамуса/гипофиза.
Нарушения миграции нейронов происходят между третьим и пятым месяцами жизни, когда нейроны не перемещаются в свои конечные места в коре головного мозга. Многие из этих аномалий представляют собой генетические пороки развития и связаны с синдромами. Большинство из них присутствуют при эпилепсии, гипотонии или задержке развития.
В случаях, когда развивается задержка нормальной радиальной миграции нейронов и глиальной ткани из перивентрикулярной зародышевой матрицы в кору, возникает гетеротопическое серое вещество. Эти места задержки миграции обнаруживаются только с помощью высокочастотных датчиков и при высоком подозрении на предполагаемую патологию, поскольку они представляются абсолютно похожими на нормальное серое вещество. Гетеротопии могут быть полосовидными или узловатыми (рис.10), а располагаться как под мягкой оболочкой мозга, так и до субэпиндемальной зоны.

Рис. 10. Гетеротопия серого вещества. Коронарная нейросограмма демонстрирует узловатые очаги серого вещества, которые охватывают стенку левого бокового желудочка.
Лиссэнцефалия – серьезная аномалия миграции, которая приводит к судорогам у всех младенцев и к смерти до 2 лет для большинства. Агирия представляет собой совершенно гладкий мозг без рельефных меток (рис.11).

Рис. 11. Лиссэнцефалия. Коронарная нейросограмма демонстрирует невыразительную кору без образования борозд.
Пахигирия приводит к образованию нескольких плоских борозд. Нормальный пренатальный мозг достаточно гладкий на 25-й неделе ГР; таким образом, точная оценка недоношенного детского гестационного возраста необходима для точного нейросонографического диагноза лиссенцефалии.
Кортикальная дисплазия с появлением признака «булыжной мостовой» называется полимикрогирией. Клинические симптомы варьируются в зависимости от места поражения. Кора выглядит слегка утолщенной и похожа на пахигирию (рис.12). Необходимо искать большие дренирующие вены, вызванные устойчивым сосудистым рисунком в мягкой и паутинной оболочках мозга эмбриона, которые покрывают аномальную кору. Идентификация этого признака должна быть подтверждена при МРТ оценке.

Рис. 12. Кортикальная дисплазия. Коронарная нейросограмма демонстрирует области с неправильными бороздами и несколько неоднородных невыразительных зон, что наблюдается при лиссэнцефалии. Пахигирия выглядит точно также.
Шизэнцефалия – это расщепление мозга, покрытое серым веществом. Дефекты могут быть большими с соответствующей задержкой развития, или небольшими с легким спазмом мышц или гипотонией. Поражения подразделяются на открытые расщелины или закрытые расщелины. Сращенные расщелины относятся к 1-му типу или шизенцефалии с закрытой расщелиной, а открытые расщелины относятся к II-му типу (рис.13).

Рис. 13. Шизенцефалия. Коронарная нейросограмма показывает шизэнцефалию II-го типа c открытой расщелиной в правой лобно-височной области. Обратите внимание на свободную связь правого бокового желудочка с экстрааксиальной жидкостью.
Связанные с этой патологией признаки включают: лентикуло-стриатную васкулопатию, отсутствие прозрачной перегородки, кортикальную дисплазию и гетеротопии серого вещества. Это поражение связано с цитомегаловирусной инфекцией.
Пролиферация и дифференциация нейронов
Стеноз водопровода, факоматоз, врожденные сосудистые пороки развития и врожденные опухоли возникают в период увеличения числа и типа клеток.
Первичный стеноз водопровода развивается в результате аномалий в дифференцировке нейронов или пролиферации периакведуктального серого вещества, но чаще возникает из-за рубцевания после сдавления, кровоизлияния или инфекции. Независимо от того, выполняется ли визуализация пренатально или постнатально, асимметричное расширение бокового и третьего желудочков при нормальном размере четвертого желудочка, является характерной отличительной чертой (см. Рис. 4). Дифференциальная диагностика от голопрозэнцефалии может быть облегчена за счет использования сосцевидного отростка (рис.14).

Рис. 14. Проекция через сосцевидный отросток. Аксиальная проекция нормального четвертого желудочка (стрелка) при доступе со стороны левого сосцевидного отростка. Обратите внимание на эхогенный червь мозжечка (V).
Для динамической оценки необходимо сохранять постоянную глубину изображения между серийными сканами в проекции через родничок для оптимального сравнения, если при этом не используется 3D-ультразвуковая техника.
Факоматозами называются синдромы аномальной пролиферации эктодермальных, мезодермальных и нейроэктодермальных компонентов, которые влияют на мозг в глобальном масштабе и приводят к развитию небольших опухолей в пределах ЦНС, мочеполовой системы и коже. Примерами являются синдром Стерджа-Вебера (энцефалотригеминальный синдром).
Пациенты имеют винный (пламенный) невус на лице, связанный с ипсилатеральной лептоменингеальной ангиомой и судорогами на первом году жизни. Синдром Стерджа-Вебера также называют энцефалотригеминальным ангиоматозом. Пораженное полушарие демонстрирует снижение объема коры головного мозга, увеличение сосудистого сплетения и серпигинозную (с волнистым краем) эхогенную плотность на периферии головного мозга (рис. 15). Самым ранним ультразвуковым признаком может быть односторонняя перивентрикулярная повышенная эхогенность.

Рис. 15. Синдром Стерджа-Вебера. Коронарная нейросограмма демонстрирует асимметрию сильвиевой борозды и клиновидную область повышенной эхогенности справа. Это лептоменингеальная ангиома (стрелка).
Туберозный склероз – это мультисистемное наследственное расстройство с множественными опухолеподобными поражениями в головном мозге, глазах, сердце, почках, легких и коже. Пре- и постнатальная сонография играет важную роль при динамическом (недорогостоящем) наблюдении за этими образованиями, поскольку многие из них могут развиться в более агрессивные опухоли, такие как субэпендимальная гигантскоклеточная астроцитома и карцинома почек. Нейросонографию зачастую проводят тогда, когда младенец имеет неспецифические неврологические симптомы. Характерные находки включают перивентрикулярные субэпендимальные узелки (обычно кальцинированные) вблизи отверстия Монро и кортикальные гамартомы, при этом все они могут проявляться в виде структур, от изо- до гиперэхогенной интенсивности, по отношению к нормальному мозгу (рис. 16).

Рис. 16. Туберозный склероз. (A) Коронарная нейросограмма новорожденного, полученная во время операции, демонстрирует аналогичную нормальному мозгу эхогенность гамартомы (H). (B) Парасагиттальная нейросограмма у другого малыша с судорогами демонстрирует субэпендимальные бугры (стрелки).
Сосудистые пороки развития (мальформации)
Сосудистые мальформации могут определяться внутриутробно в качестве увеличение синуса твердой оболочки или тромбоза, с или без расстройства развития плода, или в младенчестве при спонтанном внутричерепном кровоизлиянии или застойной сердечной недостаточности. Среди них выделяют артериовенозные пороки развития мозга, кавернозные пороки развития, артериовенозные свищи твердой мозговой оболочки, галеновые пороки развития, венозные пороки развития и венозные вариксы. Ограничения при идентификации зависят от местоположения мальформации, нейросонографического оборудования и используемых протоколов. Транскраниальная цветная дуплексная сонография в одном исследовании имела общую чувствительность 78%, но при этом остается недостаточно чувствительна, чтобы быть эффективным инструментом для скрининга. Хотя магнитно-резонансная и компьютерная томографическая артериография демонстрируют потенциал для замены методики, золотым стандартом предоперационного планирования традиционно остается ангиография. Интраоперационная сонография важна для оценки полноты резекции и идентификации питающих и дренирующих сосудов. Чувствительность исследования увеличилась с помощью применения 3D-соноангиографии. Фокальные области повышенной или уменьшенной эхогенности появляются на изображениях в серой шкале с явным фокальным увеличением васкуляризации при цветной или энергетической допплерографии (рис. 17). Для проведения исследований рекомендуем использовать аппарат от компании GE Voluson E8.

Рис. 17. Сосудистые мальформации. (A) Коронарная нейросонограмма демонстрирует небольшое увеличение эхогенности кровотока по средней линии галеновой мальформации (G). (B) Коронарная нейросограмма с дуплексным допплеровским трассированием демонстрирует бурный кровоток, как признак галеновой мальформации. (C) Парасагитальная нейросограмма через височную ямку другого пациента демонстрирует венозную ангиому, в виде очаговой области повышенной эхогенности (стрелки).



